近期,广州医科大附属第三医院和美国得克萨斯大学西南医学中心合作,利用基于质谱的蛋白质组学鉴定了参与铁死亡的一个关键膜蛋白HPCAL1,并深入研究了其作用机制及潜在的靶向治疗可能性。研究成果以“Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis”为题,发表在Autophagy(IF:13.391)上。

研究结果
1. HPCAL1 对铁死亡是必须的
铁死亡表现为细胞器和细胞膜的脂质过氧化,但关键的膜调控蛋白仍然未知。研究者使用铁死亡激活剂RSL3(一种谷胱甘肽过氧化物酶 4 (GPX4) 的抑制剂,诱导铁死亡)分别处理人纤维肉瘤细胞HT-1080和人非小细胞肺癌细胞Calu-1,随后分离细胞膜组分并进行基于质谱的蛋白质组学检测,以期寻找关键的铁死亡膜调控蛋白。
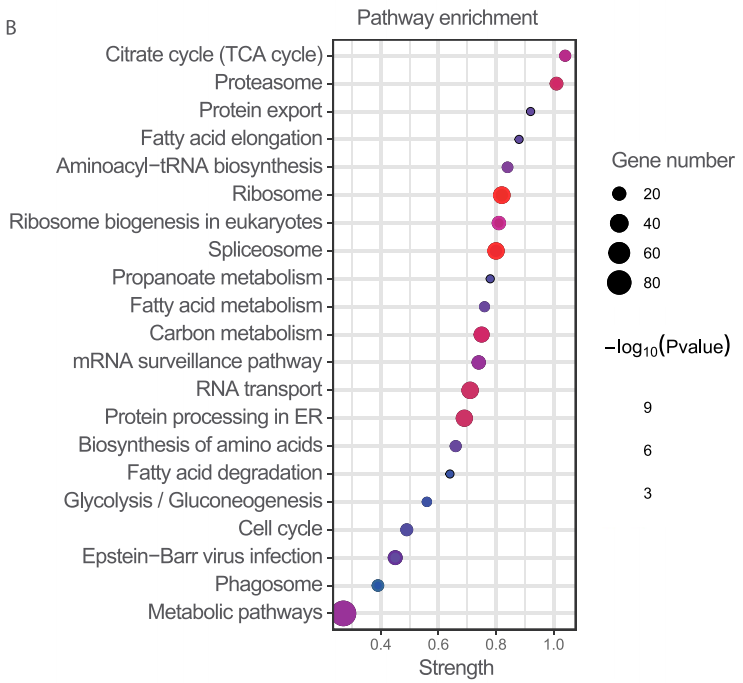
细胞膜组分的蛋白质组学共鉴定了752个膜蛋白,这些膜蛋白的通路富集分析显示,三羧酸循环代谢是铁死亡中最受影响的通路,与此前报道的线粒体三羧酸循环和脂质代谢参与铁死亡的报道相一致。
在铁死亡激活剂RSL3处理后的HT-1080和Calu-1中有16个共有上调蛋白,包括MAGOH、SPTAN1、OSTM1、HPCAL1、CPT2、FKBP1A、EBNA1BP2、FBL、PRDX3、H1-3/HIST1H1D、ZYX、 UBE2M、ANXA7、EIF4G1、HNRNPUL1和PRKAR1A。随后,研究者在两轮实验中分别使用靶向这些16个蛋白基因的siRNA处理HT-1080和Calu-1细胞,最终仅有HPCAL1 siRNA能同时降低RSL3诱导的细胞死亡。因此,研究者锁定HPCAL1进行后续探索。
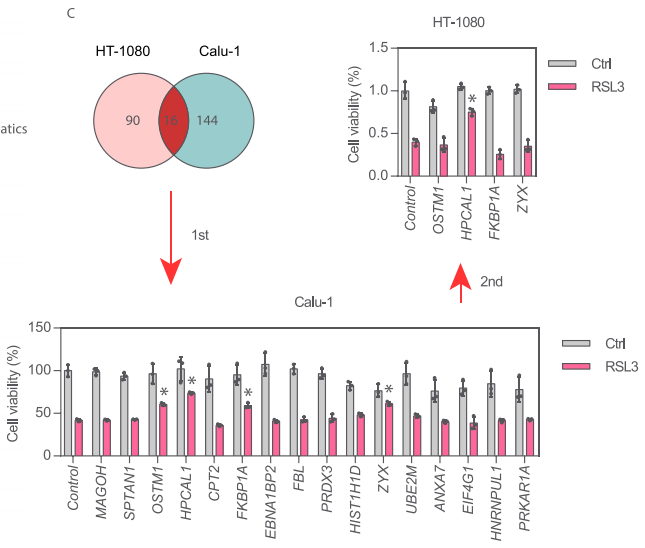
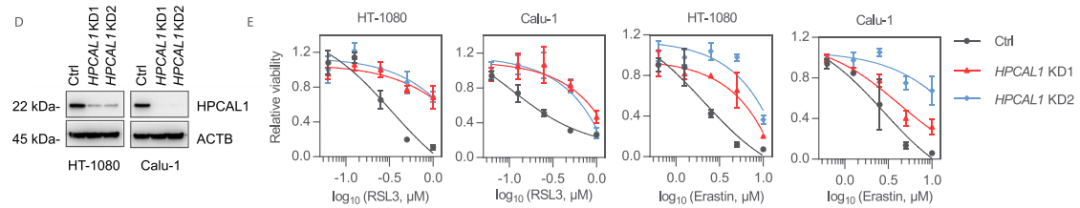
为了进一步确认HPCAL1在促进铁死亡中的作用,研究者构建了能稳定敲低HPCAL1的两个特异性shRNA。与HPCAL1 siRNA实验结果一致,HPCAL1 shRNA抑制了RSL3或erastin(另一种铁死亡诱导剂)诱导的HT-1080和Calu-1细胞生长抑制。
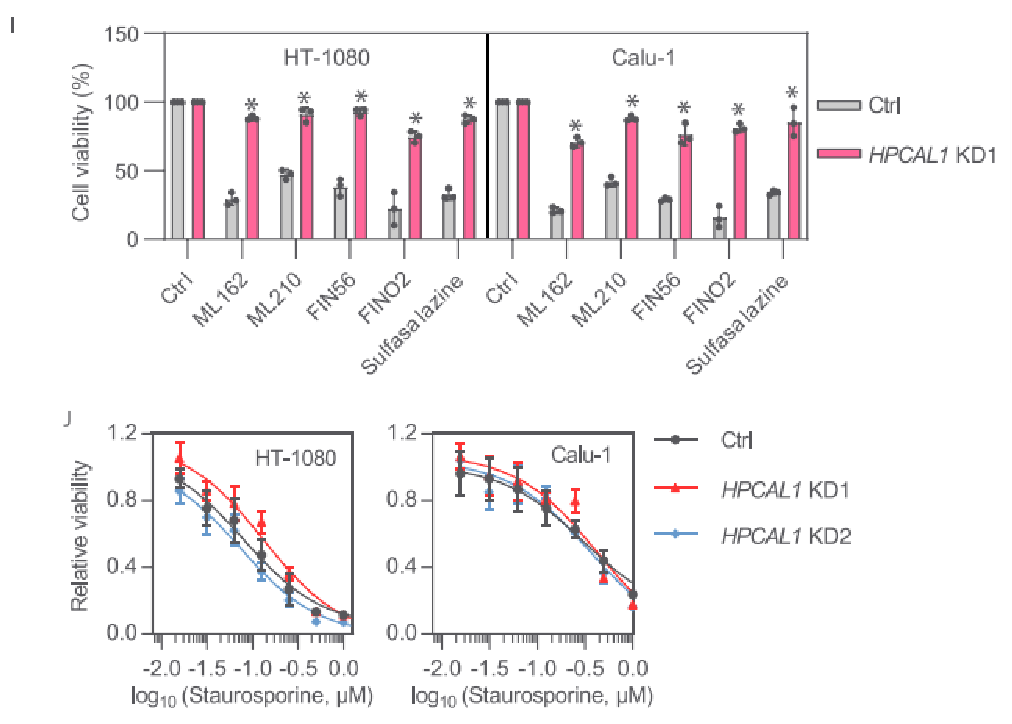
随后的研究也表明,HPCAL1缺失不影响细胞膜的完整性。此外,HPCAL1缺失将显著降低脂质过氧化,因此HPCAL1可能是铁死亡中脂质过氧化的正调节剂。研究者继续使用其他铁死亡诱导剂,包括ML162、ML210、FIN56、FINO2和sulfasalazine处理HT-1080和Calu-1细胞,结果均证实了HPCAL1的促铁死亡作用。而形成鲜明对比的是,细胞凋亡诱导剂staurosporine (STS)导致的细胞死亡并不受到HPCAL1敲低的影响。这一结果表明,HPCAL1是选择性促进铁死亡而非细胞凋亡。
研究者随后对HPCAL1进行过表达,过表达后细胞对铁死亡诱导剂RSL3或erastin的处理更加敏感。此外,在HPCAL1敲低的细胞中重新稳定过表达HPCAL1后,细胞恢复了对RSL3或erastin处理的敏感性,这种作用会被铁死亡抑制剂liproxstatin-1抑制,而不受细胞凋亡抑制剂Z-VAD-FMK的影响。
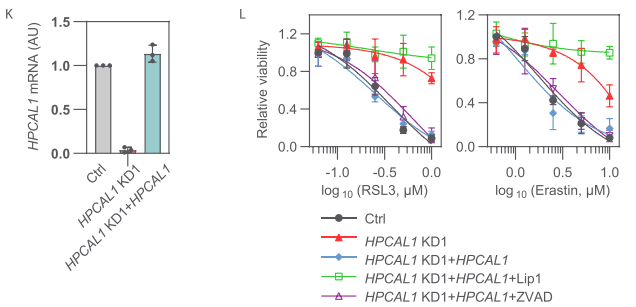
HPCAL1最开始是在视网膜和脑组织中被检测到的,而该蛋白其实广泛表达于各种组织和细胞中。研究者在其他种类的人癌细胞系中使用shRNA敲低HPCAL1,结果显示HPCAL1敲低均可以抑制RSL3或erastin诱导的细胞死亡。综合以上研究表明,HPCAL1介导广泛的铁死亡。
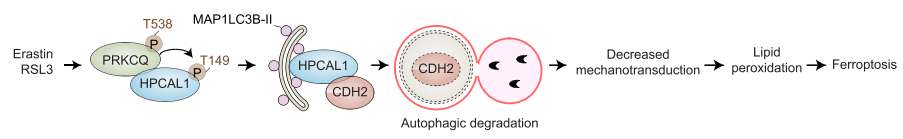
2. HPCAL1介导的CDH2抑制能促进铁死亡
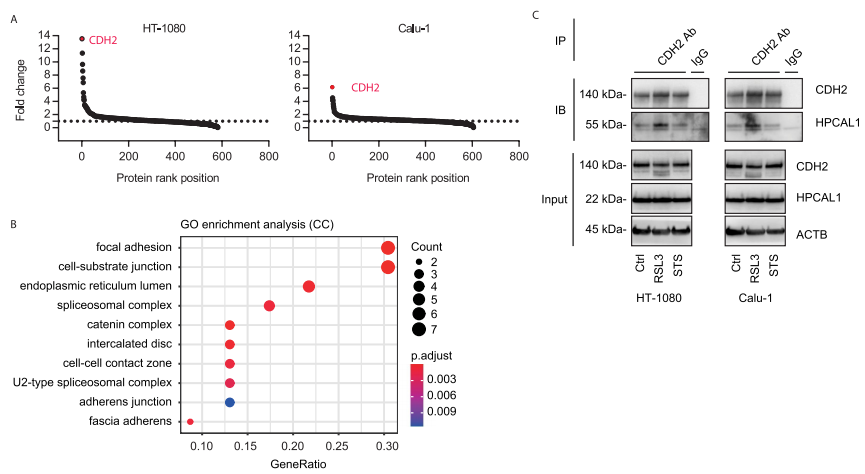
研究者进一步探索了HPCAL1促进铁死亡的作用机制。首先,研究者通过实验排除了钙离子参与HPCAL1介导的铁死亡以及HPCAL1在铁死亡中催化脂质通透性/氧化。随后,研究者检测HPCAL1是否与铁死亡相关蛋白结合。通过HPCAL1的CO-IP MS实验,共发现在RSL3处理和未处理的HT-1080和Calu-1细胞中,与HPCAL1结合的蛋白有600多个。GO功能富集分析显示粘着斑和细胞-底物接合是最显著富集的条目。
研究者关注其中一个在RSL3处理后与HPCAL1结合变化倍数最大的蛋白CDH2。随后,研究者分别用RSL3(铁死亡激活剂)和STS(细胞凋亡诱导剂)处理细胞,并进行CDH2的CO-IP。结果表明,在铁死亡状态下而非细胞凋亡状态下,CDH2与HPCAL1的结合加强。该结果显示了HPACAL1-CDH2复合体在铁死亡中的特殊作用。
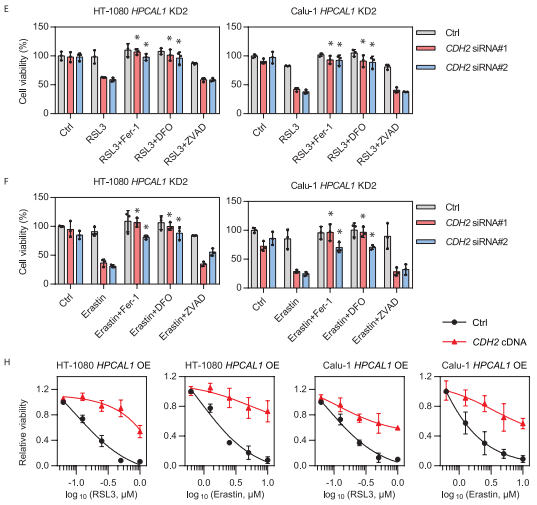
接下来的研究显示,当CDH2被敲低后,能恢复HPCAL1敲低细胞对RSL3或erastin的敏感性,而这种作用可以被铁死亡抑制剂DFO而非细胞凋亡抑制剂Z-VAD-FMK逆转。而在HPCAL1过表达的细胞中继续过表达CDH2则会限制RSL3或erastin的细胞杀伤活性。综合以上结果表明,HPCAL1通过劫持抗铁死亡CDH2蛋白来促进铁死亡。
3. HPCAL1介导CDH2降解
紧接着研究者探究HPCAL1-CDH2复合体的形成是如何抑制CDH2的抗铁死亡作用。首先发现,在RSL3或erastin处理后,细胞中的CDH2蛋白表达显著下调,而细胞凋亡诱导剂STS则不影响CDH2的表达。研究者随后明确,CDH2的降解是铁死亡过程中的自噬介导的,在铁死亡过程中自噬流增强。
自噬受体一般会将吞噬泡和自噬小体上的MAP1LC3B-II和要降解的目标分子进行连接,因此研究者检测了HPCAL1和MAP1LC3B-II的膜脂质定位。WB显示在RSL3诱导的铁死亡条件下,细胞膜组分中的HPCAL1和MAP1LC3B-II表达量均上升。HPCAL1可以与磷脂酰肌醇结合,这与此前报道的磷脂酰肌醇是自噬小体形成和成熟中关键的协调分子相一致。此外,CO-IP实验也证实,在RSL3诱导的铁死亡条件下,膜组分存在HPCAL1、CDH2和MAP1LC3B-II的结合。
下一步,研究者的实验表明:(1)在铁死亡中HPCAL1选择性地介导CDH2蛋白的自噬降解。(2)HPCAL1上预测到的两个LIR(LC3互作区)motif :DEFFKKI (aa 46-51) 和AIYKMV (aa 127-132)。HPCAL1突变实验表明, Δ127-132缺失不影响HPCAL1与LC3的结合,而Δ46-51缺失则影响HPCAL1与LC3的结合。此外,Δ46-51缺失抑制RSL3或erastin诱导的CDH2降解。
4. PRKCQ介导的HPCAL1磷酸化促进CDH2的降解
研究者继续探索了HPCAL1磷酸化是否以及如何调控铁死亡中的CDH2降解。研究者通过实验初步确认PRKC(蛋白激酶C)的底物选择性参与铁死亡相关的自噬。随后,研究者明确HPCAL1是PRKC的潜在底物。生信预测显示,PRKC可能磷酸化HPCAL1 149位苏氨酸(Thr)的磷酸化。功能实验表明,过表达野生型HPCAL1(Thr149)增强CDH2降解,而过表达磷酸化突变(T149A,149位苏氨酸突变为丙氨酸)则较少CDH2的降解。两个PRKC抑制剂则抑制RSL3或erastin诱导的铁死亡。以上结果表明,PRKC依赖的HPCAL1磷酸化介导了铁死亡中CDH2的降解。此外,研究者的实验表明,是PRKC家族中的PRKCQ Thr538的磷酸化使的PRKCQ激活,参与了铁死亡。
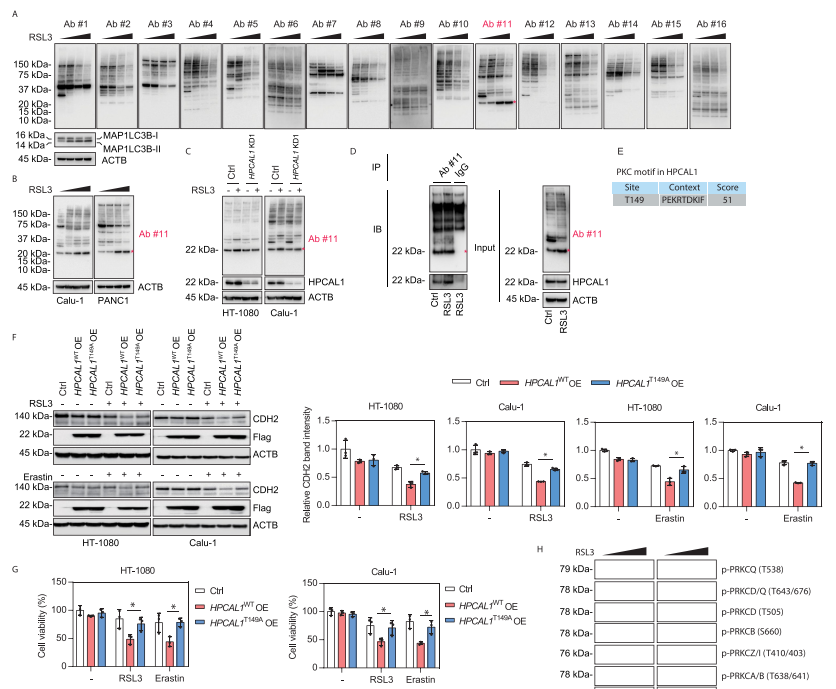
接下来,研究者检测了PRKCQ在铁死亡中的作用。结果表明,铁死亡激活剂RSL3而非凋亡诱导剂STS能增强PRKCQ和HPCAL1的结合。PRKCQ敲低限制了RSL3或erastin诱导的癌细胞生长抑制。PRKCQ敲低保护细胞免受细胞死亡、脂质过氧化和CDH2的自噬降解。此外,在细胞缺失HPCAL1和CDH2的情况下,敲低PRKCQ不影响细胞对铁死亡敏感性。这表明,PRKCQ是铁死亡的上游信号。
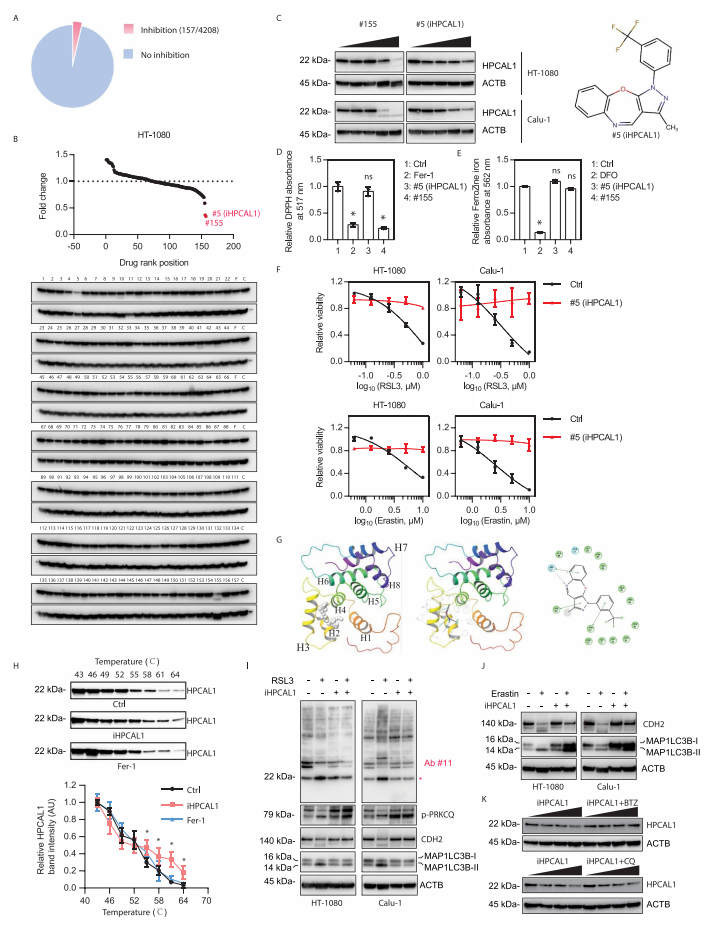
5. 鉴定HPCAL1抑制剂(iHPCAL1)可作为铁死亡的抑制剂
研究者从4208个无明确靶点的小分子中筛选新的HPCAL1抑制剂。实验发现,有157个化合物能抑制RSL3诱导的细胞死亡。接下来,研究者检测这157个化合物中哪些可以降低HPCAL1的蛋白表达水平。结果发现,#5和#155号化合物可以显著抑制HPCAL1蛋白表达。随后,研究者关注既不是抗氧化剂也无铁螯合剂活性的#5号化合物,并将之命名为iHPCAL1。分子对接实验(molecular docking assay)显示iHPCAL1可以与HPCAL1结合。
细胞热迁移分析(cellular thermal shift assay)证实iHPCAL1延缓热诱导的HPCAL1蛋白降解,表明iHPCAL1直接与HPCAL1相互作用。功能分析表明iHPCAL1不能影响PRKCQ的磷酸化,但可以抑制HPCAL1的磷酸化以及RSL3或erastin诱导的CDH2的自噬降解。这些结果表明,iHPCAL1诱导的HPCAL1磷酸化和功能的抑制发生在PRKCQ 磷酸化的下游。随后的研究表明硼替佐米(泛素-蛋白酶体抑制剂),而不是氯喹(自噬抑制剂),可以阻止iHPCAL1诱导的HPCAL1蛋白降解。这些结果表明iHPCAL1通过蛋白酶体途径刺激HPCAL1的降解,从而限制了铁死亡。
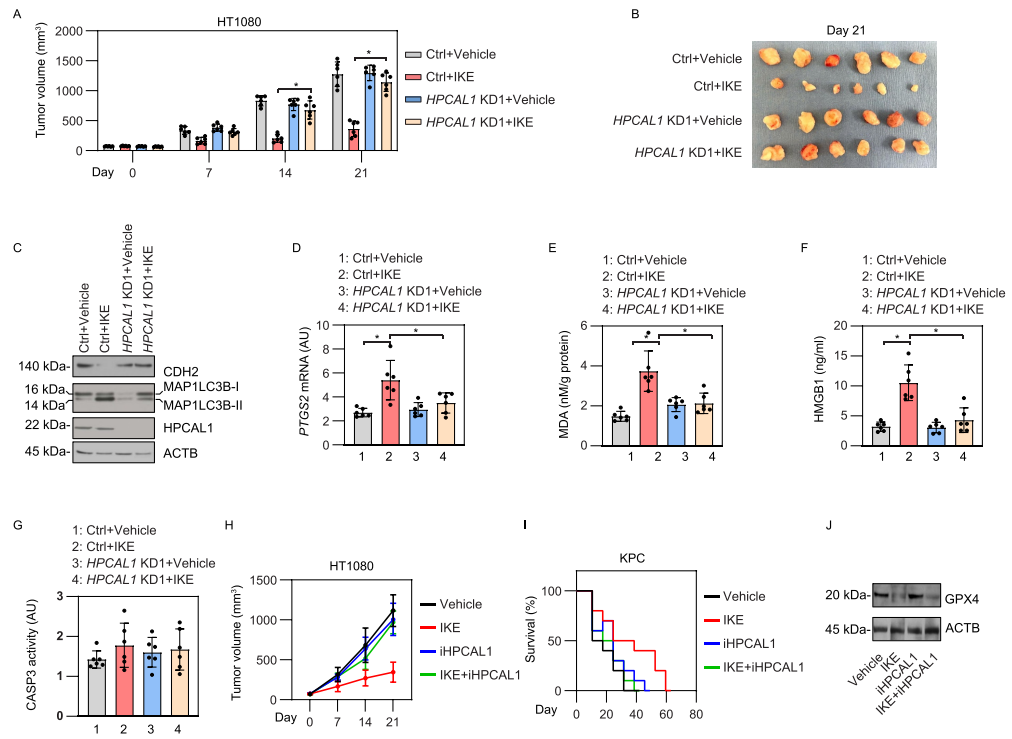
6. 体内实验检测HPCAL1介导的铁死亡响应
Imidazole ketone erastin (IKE)是一种具有更好效力和代谢稳定性的erastin类似物,适用于在动物研究评估铁死亡对肿瘤抑制的影响。研究者构建了HPCAL1敲低的HT-1080癌细胞异种移植瘤裸鼠模型,并发现敲低HPCAL1限制了IKE的肿瘤抑制作用。HPCAL1敲低组与对照组相比,CDH2蛋白水平升高,而MAP1LC3B-II降低。铁死亡生物标志物的定量结果以及血清中HMGB1的浓度进一步支持HPCAL1促进了IKE诱导的体内铁死亡的假设。同样地,iHPCAL1在异种移植瘤模型中抑制IKE的抗癌活性,而对IKE诱导的GPX4降解无显著影响。这些动物体内研究与体外研究一致,表明HPCAL1是铁死亡的正调节因子。
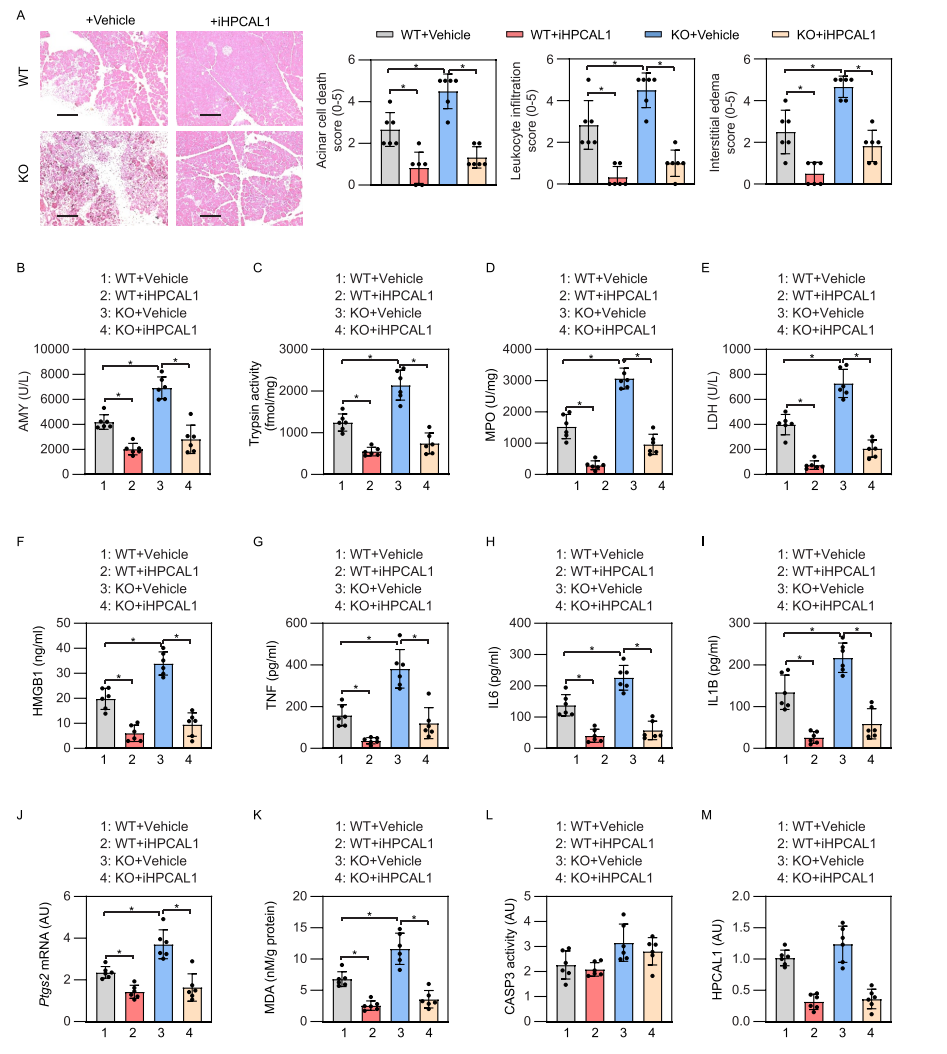
随后,研究者利用蛙皮素(cerulein)诱导胰腺条件性敲除GPX4的小鼠发生急性胰腺炎,并检测iHPCAL1是否有保护作用。结果显示,iHPCAL1能很好地改善急性胰腺炎中的各项生化指标,具有显著的保护作用。与预期一致,在使用iHPCAL1后,HPCAL1蛋白水平下调。综合以上结果表明,HPCAL1参与了铁死亡诱导的急性胰腺炎。
研究总结
研究者利用定量蛋白质组学筛选到了一个参与铁死亡的膜蛋白HPCAL1。HPCAL1作为一种特异性自噬受体,在铁死亡过程中选择性降解CDH2(钙粘蛋白2)。CDH2耗竭又进一步通过降低膜张力和促进脂质过氧化而增加铁死亡的易感性。深入的机制研究发现,CDH2的自噬降解需要PRKCQ(蛋白激酶C theta)介导的HPCAL1在Thr149上的磷酸化,以及位于46-51位氨基酸之间的非经典LC3互作区域motif。最后,研究者从4208个小分子化合物的药物筛选中确定了一种抑制HPCAL1表达的铁死亡抑制剂。在小鼠模型中,HPCAL1的抑制阻止了铁死亡诱导的肿瘤抑制和胰腺炎。研究者的发现为自噬依赖细胞死亡的机制提供了见解,并为靶向HPCAL1以预防致病性铁死亡提供了可能性。
吉凯基因凭借多年在靶标筛选及验证服务领域的技术积累,建立的标准化 、工程化 、系统化的GRP平台,为中国研究型医生提供科研服务,加快科研成果转化。其中,多组学平台包含蛋白质组学平台和高通量测序平台:
·蛋白质组学平台拥有多台timsTOF Pro、Exploris 480高精度质谱仪,专业的Spectronaut Plusar、Mascot等分析软件,提供专业的4D、DIA、TMT、PRM、磷酸化修饰组、olink蛋白质组等检测服务,强大的机器学习算法、IPA分析、蛋白基因组分析服务,系统的生物标志物、分子分型、药物靶点、基因功能研究等解决方案,真正让广大研究型医生的科研工作更省心、更省力、更高效;
·高通量测序平台分为常规测序服务和单细胞测序服务:单细胞测序拥有10x和BD两个平台,提供单细胞RNA-seq、单细胞核测序、单细胞混样RNA-seq、单细胞TCR/BCR、单细胞(RNA+ATAC)、空间转录组测序等服务;常规测序服务提供meRIP-seq(m6A/m1A/m7G/m5C 等RNA甲基化修饰测序)、acRIP-seq(ac4C RNA乙酰化修饰测序)、ATAC-seq、Ribo-seq(翻译组测序) 、mRNA/miRNA/LncRNA/circRNA-seq、全转录组测序(两文库/三文库)、外泌体miRNA/LncRNA-seq、WGS/WES、WGBS、RRBS、BSAS等服务。