动植物基因组从头测序
产品名称: 动植物基因组从头测序
英文名称: Whole-genome de novo sequencing of plants and animalsand plants genome
产品编号:
产品价格: 0
产品产地: null
品牌商标: null
更新时间: null
使用范围: null
- 联系人 :
- 地址 : 上海市徐汇区银都路218号聚科生物园区2号楼
- 邮编 : 200231
- 所在区域 : 上海
- 电话 : 186****3700 点击查看
- 传真 : 点击查看
- 邮箱 : market@personalbio.cn;lif@personalbio.cn
动植物基因从头测序
基因组从头测序也叫de novo测序,是指对基因组序列未知或没有近源物种基因组信息某个物种对其不同长度基因组DNA片段及其文库进行序列测定,然后用生物信息学方法进行拼接、组装和注释,从而获得该物种完整的基因组序列图谱。
一、技术路线
动植物基因组de novo测序的主要策略:一是对短片段Shotgun文库(300-1000 bp)进行深度测序,确保序列覆盖度和测序准确性,获得基因组基本序列信息;二是构建较长插入片度的mate pair文库(3kb、8 kb、10 kb、20 kb)并测序,确定短片段序列间的相对位置,通过拼接组装获得基因组序列框架;三是通过PCR扩增技术获得序列间断开部分(gap)DNA片段并进行一代测序,从而获得完整基因组序列。在此基础上通过生物信息学方法进行基因注释和分析。
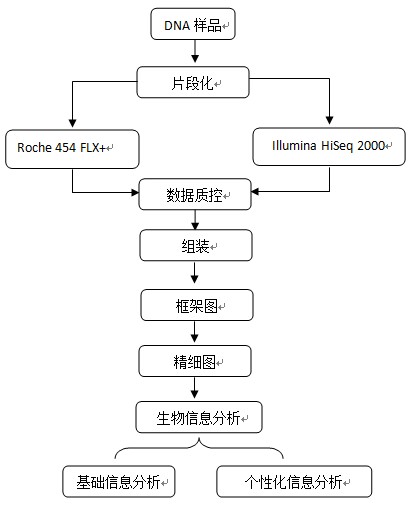
推荐平台:Roche 454 FLX+数据为主,结合Illumina HiSeq 2000和ABI 3730XL 数据。
二、生物信息分析
1. 原始数据整理、过滤及质量评估
2. 基因组序列拼装与分析:
l 基因组序列拼装
l 基因组拼装效果评估 (contig N50、scaffold N50、genome size and GC% )
l 比较基因组分析(进化速率分析、亲缘关系分析、SNPs、CNV、共线性以及重复片段分析)
3. 功能元件分析:
l 蛋白编码基因预测
l 非编码 RNA预测
l 蛋白编码基因的功能注释
l 蛋白编码基因的KOG和 GO 注释
l 蛋白编码基因的代谢途径注释(KEGG pathway)
4. 根据客户需求进行个性化分析
三、样品要求
1. DNA样品:浓度≥200 ng/μl,总量≥500 μg,OD 260/280值应在1.8-2.0之间,电泳检测无明显RNA条带,基因组条带清晰、完整,主带应在100 kb以上。若样品中有多糖、糖蛋白的残留,对打断DNA样品带来非常大的困难,且很难去除,因此特别要求所提供的样品一定要将多糖或糖蛋白去除干净。
2. 动物样品:样品最好来自纯系,对于一般物种应挑选肝脏、肾脏、血液等组织取样,对于珍贵物种请提供耳样、毛发(带毛根)等脂肪含量较少的组织进行取样。为了减少个体差异对后续拼接产生的影响,尽量从同一个个体中取样。若物种体积较小,从一个个体中提取的DNA量不能满足测序实验所需,在保证量的前提下,应尽量减少采样个体的数量。
3. 植物样品:样品最好来自纯合体或单倍体,需为黑暗无菌条件下培养的黄化苗或组织样品。
四、经典案例
案例1:猩猩全基因组测序
背景:红毛猩猩(Pongo pygmaeus)属猩猩科,是一种非常珍稀的灵长类动物,目前只在婆罗洲低地和苏门答腊洲有少量存活,全世界仅剩下不到3万只。红毛猩猩与大猩猩、黑猩猩一起被称为“人类最直系的亲属”。
目的:对6个苏门答腊猩猩和5个婆罗洲猩猩进行测序,解析苏门答腊猩猩和婆罗洲猩猩的遗传多样性,并研究类人猿包括人类的进化过程。

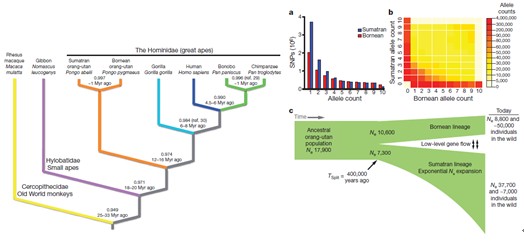
结果:测序得到总数据量约为300 Gb。经过比对发现,苏门答腊猩猩和婆罗洲猩猩在40万年前才进化为不同的物种,比前期估计要晚60万年。红毛猩猩起源于约1200万至1600万年前,而人类和黑猩猩起源于约500万至600万年前,与人类和黑猩猩相比,红毛猩猩进化的时间更久,但对这三种猿的基因组进行比对发现,红毛猩猩DNA的变异程度远远小于人类和黑猩猩。
[ Locke DP, Hillier LW, Warren WC, et al. Comparative and demographic analysis of orang-utan genomes. Nature, 2011, 469: 529–533 ] 案例2:番茄全基因组测序 背景:番茄(Solanum lycopersicum)属茄科作物,是主要的栽培作物,并且是研究果实发育的模式作物。关于番茄的各种特性,包括味道,天然抗虫性,以及营养成分已经有许多研究,但是控制这些性状的基因机理未知,对番茄全基组进行测序可以了解这些基因的表达和调控模式,并为番茄的育种提供指导。 目的:运用Roche 454、Illumina和ABI 3730XL平台对番茄全基因组进行测序,通过与已测序完成植物物种的比较基因组学分析,研究番茄的进化事件,从而了解功能基因进化过程的信息。 结果:基因组大小约为900 Mb,共鉴定出约34727个基因,其中97.4%的基因已精确定位到染色体上。进化分析表明,西红柿基因组在进化过程中有两次基因组的三倍复制事件(triplicate),使基因家族产生了特异控制果实发育及营养品质的新成员。番茄全基因组测序工作的完成有利于进一步改善番茄和其他作物的产量抗病性、风味和颜色等。 [ Sato S, Tabata S, Hirakawa H, et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature, 2012, 485: 635-641 ] 案例3:帝王蝶全基因组测序 背景:黑脉金斑蝶(Danaus plexippus),俗称帝王蝶,是栖息在北美地区的一种色彩斑斓、身体硕大的蝴蝶。它是地球上唯一进行季节性长途迁徙的蝴蝶,整个一次迁徙过程往往需要5代蝴蝶来完成,这种迁徙习性,被誉为世界一大自然奇观。 目的:对帝王蝶全基因组进行测序,了解它长途迁徙的遗传和分子机制。 结果:帝王蝶基因组大小273 Mb,含有16,866个蛋白编码基因;比较基因组学发现帝王蝶和家蚕具有相对较近的亲缘关系。分析了负责视图、生物钟、方向性飞行的基因,同时揭示了保幼激素合成所需的一整套基因。这些都有助于帮助我们更加深入地了解帝王蝶长途迁移的遗传和分子机制。 [ Zhan S, Merlin C, Boore JL, et al. The Monarch Butterfly Genome Yields Insights into Long-Distance Migration. Cell, 2011, 147(5): 1171-1185 ]

五、常见问题解答
1. Q:基因组框架图和精细图有什么不同?
A:框架图能覆盖基因组常染色体区域90%,覆盖基因区域95%,contig N50达到5 kb,scaffold N50达到20 kb,单碱基错误率在十万分之一以下。
精细图能覆盖基因组常染色体区域95%,覆盖基因区域98%,contig N50达到20 kb,scaffold N50达到300 kb,单碱基错误率在十万分之一以下。
2. Q:动植物基因组从头测序时为什么需要构建不同类型的文库?
A:因为动植物基因组大、复杂度高,且存在大量的重复区域,因此需要制备不同梯度的测序文库,进行双末端测序,使得在拼接中能够跳过大范围的重复区,从而避免了基因组中重复序列造成的错拼,提高了拼接的质量;同时结合BAC或Fosmid文库以及多种测序平台的综合运用,保证了测序的准确性和基因组的完整性,高效经济的完成高等动植物的基因组图谱绘制。
3. Q:在动植物基因组从头测序中,不同测序平台分别有什么优势?
A:Roche 454 FLX+具有长读长能力,单条序列最长能达到800-1000 bp,在该平台上能够构建较长插入片度的mate pair文库,如8 kb、10 kb、20 kb等,可以为基因组的组装提供高质量的框架。Illumina HiSeq 2000平台优势是测序数据量大,成本低,可以为基因组测序提供高覆盖率的数据。根据物种基因组的特点及复杂程度,派森诺生物会科学地设计测序实验方案,合理地将两个平台搭配在一起使用,既能解决复杂基因组的测序难题,保证基因组测序的质量,同时也能兼顾实验项目的经济性。